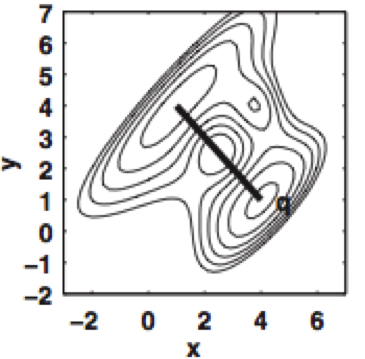
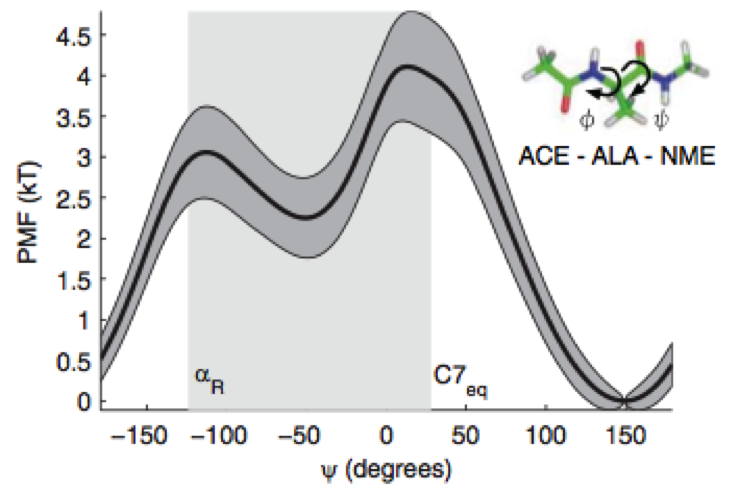
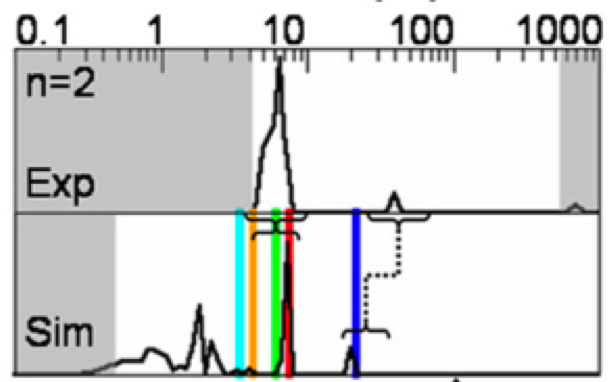
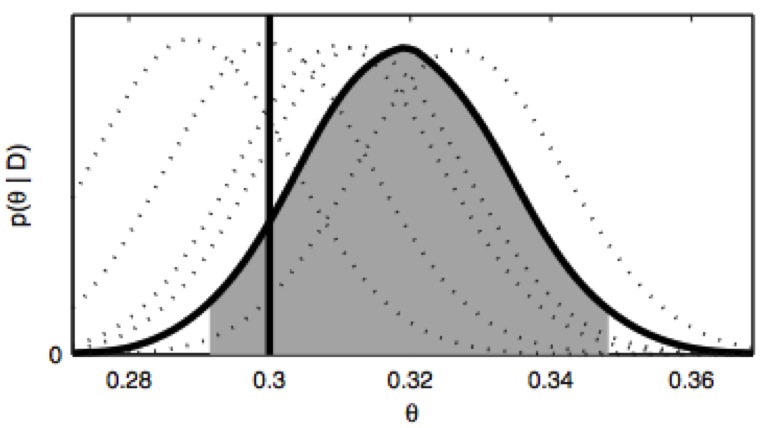
Nonequilibrium candidate Monte Carlo is an efficient tool for equilibrium simulation
/
Jerome P. Nilmeier, Gavin E. Crooks, David D. L. Minh, and John D. Chodera.
Proc. Natl. Acad. Sci. USA 108:E1009, 2011. [DOI] [PDF]
We present a significant generalization of Monte Carlo methods that provide an enormously useful tool for enhancing the efficiency of molecular simulations and enabling molecular design.
Keywords: NCMC; Monte Carlo; Metropolis-Hastings; acceptance rates; molecular dynamics