
SEARCH
ARTICLE TYPES

OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials
/Eastman P, Galvelis R, Peláez RP, Abreu CRA, Farr SE, Gallicchio E, Gorenko A, Henry MH, Hu F, Huang J, Krämer A, Michel J, Mitchell J, Pande VS, Rodrigues JPGLM, Rodriguez-Guerra J, Simmonett AC, Swails J, Turner P, Wang Y, Zhang I, Chodera JD, De Fabritiis G, Markland TE
Journal of Physical Chemistry B [DOI] [website] [code]
We present OpenMM 8, which includes GPU-accelerated support for simulating hybrid ML/MM systems that use machine learning (ML) potentials to achieve high accuracy with minimal loss in speed.
Open science discovery of potent noncovalent SARS-CoV-2 main protease inhibitors
/Boby ML, Fearon D, Ferla M, Filep M, Robinson MC, The COVID Moonshot Consortium, Chodera JD, Lee A, London N, von Delft F.
Science 382:eabo7201, 2023 [DOI] [ready to use data]
We report the discovery of a new oral antiviral non-covalent SARS-CoV-2 main protease inhibitor developed by the COVID Moonshot, a global open science collaboration leveraging free energy calculations on Folding@home and ML-accelerated synthesis planning, now in accelerated preclinical studies funded by an $11M grant from the WHO ACT-A program via the Wellcome Trust. We are currently in discussions with generics manufacturers about partnering with us throughout clinical trials to ensure we can scale up production for global equitable and affordable access once approved by regulatory agencies.
Identifying and Overcoming the Sampling Challenges in Relative Binding Free Energy Calculations of a Model Protein:Protein Complex
/
Zhang I, Rufa DA, Pulido I, Henry MM, Rosen LE, Hauser K, Singh S, Chodera JD
Journal of Chemical Theory and Computation 19:4863, 2023
We assess what is required for alchemical free energy calculations to be able to make high-quality predictions of the impact of interfacial mutations on protein-protein binding.
Development and benchmarking of Open Force Field 2.0.0---the Sage small molecule force field
/
Boothroyd S, Behara PK, Madin OC, Hahn DF, Jang H, Gapsys V, Wagner JR, Horton JT, Dotson DL, Thompson MW, Maat J, Gokey T, Wang L-P, Cole DJ, Gilson MK, Chodera JD, Bayly CI, Shirts MR, Mobley DL
Journal of Chemical Theory and Computation 19:3251, 2023 [DOI] [chemRxiv] [GitHub] [examples]
We present a new generation of small molecule force field for molecular design from the Open Force Field Initiative fit to both quantum chemical and experimental liquid mixture data
MEN1 mutations mediate clinical resistance to menin inhibition
/
Perner F, Stein EM, Wenge DV, Singh S, Kim J, Apazidis A, Rahnamoun H, Anand D, Marinaccio C, Hatton C, Wen Y, Stone RM, Schaller D, Mowla S, Xiao W, Gamlen HA, Stonestrom AJ, Persaud S, Ener E, Cutler JA, Doench JG, McGeehan GM, Volkamer A, Chodera JD, Nowak RP, Fischer ES, Levine RL, Armstrong SA, Cai SF
Nature 615:913, 2023 [DOI]
We describe how mutants that confer therapeutic resistance to menin inhibition impact small molecule binding but not interactions with the natural ligand MLL1.
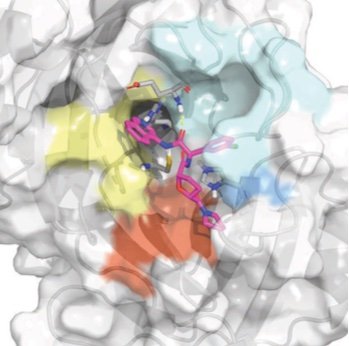
Turning high-throughput structural biology into predictive inhibitor design
/
Saar KL, McCorkindale W, Fearon D, Boby M, Barr H, Ben-Shmuel A, COVID Moonshot Consortium, London N, von Delft F, Chodera JD, Lee AA
PNAS 120:e2214168120, 2023 [DOI]
We demonstrate how potent inhibitors can be predicted from high-throughput structural biology, demonstrating this approach against the SARS-CoV-2 main viral protease (Mpro).
SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials
/
Eastman P, Behara PK, Dotson DL, Galvelis R, Herr JE, Horton JT, Mao Y, Chodera JD, Pritchard BP, Wang Y, De Fabritiis G, and Markland TE
Scientific Data 10:11, 2023 [DOI]
To remedy the lack of large, open quantum chemical datasets for training accurate general machine learning potentials and molecular mechanics force fields for druglike small molecules and biomolecules, we produce the open SPICE dataset, and show how it can be used to build extremely accurate machine learning potentials.
Improving force field accuracy by training against condensed-phase mixture properties
/
Boothroyd S, Madin OC, Mobley DL, Wang L-P, Chodera JD, and Shirts MR
Journal of Chemical Theory and Computation 18:3577, 2022 [DOI] [GitHub]
We use a new automated framework for physical property evaluation and fitting to show how molecular mechanics force fields can be systematically improved by fitting to condensed phase properties.
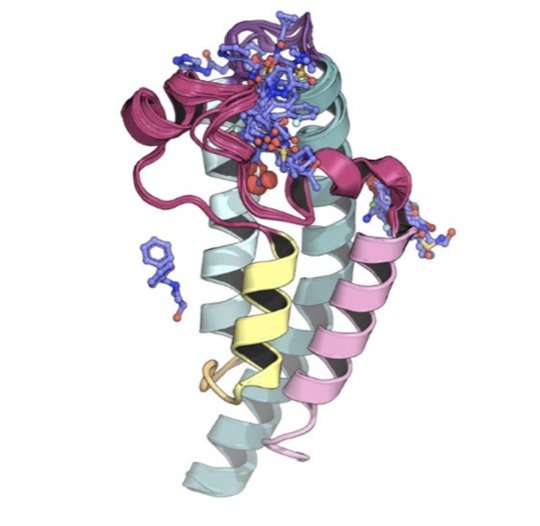
SAMPL7 protein-ligand challenge: A community-wide evaluation of computational methods against fragment screening and pose-prediction
/
Grosjean H, Isik M, Aimon A, Mobley D, Chodera JD, von Delft F, and Biggin PC
Journal of Computer-Aided Molecular Design 36:291, 2022 [DOI]
We field a blind community challenge to assess how well state of the art computational chemistry methods can predict the binding modes of small druglike fragments to a protein target for which no chemical matter is known, PHIP2, using fragment screening at the Diamond Light Source.
CACHE (Critical Assessment of Computational Hit-finding Experiments): A public-private partnership benchmarking initiative to enable the development of computational methods for hit-finding
/
Ackloo S, Al-awar R, Amaro RE, Arrowsmith CH, Azevedo H, Batey RA, Bengio Y, Betz UAK, Bologa CG, Chodera JD, Cornell WD, Dunham I, Ecker GF, Edfeldt K, Edwards AM, Gilsom MK, Gordijo CR, Hessler G, Hillisch A, Hogner A, Irwin JJ, Jansen JM, Kuhn D, Leach AR, Lee AA, Lessel U, Moult J, Muegge I, Oprea TI, Perry BG, Riley, Singh Saikantendu K, Santhakumar V, Schapira M, Scholten C, Todd MH, Vedadi M, Volkamer A, and Wilson TM
Nature Reviews Chemistry 6:287, 2022 [DOI]
We describe CACHE: A new public-private partnership that aims to transform computer-aided drug discovery much the way that CASP transformed protein structure prediction into a reproducible, accurate engineering discipline.
INK4 tumor suppressor proteins mediate resistance to CDK4/6 kinase inhibitors
/
Li Q, Jiang B, Guo J, Shao H, Del Priore IS, Chang Q, Kudo R, Li Z, Razavi P, Liu B, Boghossian AS, Rees MG, Ronan MM, Roth JA, Donovan KA, Palafox M, Reis-Filho JS, de Stanchina E, Fischer ES, Rosen N, Serra V, Koff A, Chodera JD, Gray NS, and Chandardlapaty S
Cancer Discovery} 12:356, 2022 [DOI]
We demonstrate CDK6 causes drug resistance by binding INK4 proteins, and develop bifunctional degraders conjugating palbociclib with E3 ligands to overcome this mechanism of resistance.
Quantum chemistry common driver and databases (QCDB) and quantum chemistry engine (QCEngine): Automation and interoperability among computational chemistry programs
/
Smith DGA, Lolinco AT, Glick ZL, Lee J, Alenaizan A, Barnes TA, Borca CH, Di Remigio R, Dotson DL, Ehlert S, Heide AG, Herbst MF, Hermann J, Hicks CB, Horton JT, Hurtado AG, Kraus P, Kruse P, Lee SJR, Misiewicz JP, Naden LN, Ramezanghorbani F, Scheurer M, Shriber JB, Simmonett AC, Steinmetzer J, Wagner JR, Ward L, Welborn M, Altarawy D, Anwar J, Chodera JD, Dreuw A, Kulik HJ, Liu F, Martinez TJ, Matthews DA, Schaefer III HF, Sponer J, Turney JM, Wang L-P, De Silva N, King RA, Stanton JF, Gordon MS, Windus TL, Sherrill CD, Burns LA
Journal of Chemical Physics} 155:204801, 2021 [DOI]
We describe a new community-wide approach to interoperability for quantum chemistry packages that will enable large-scale applications such as next-generation machine learning for chemistry and automated force field construction for drug discovery.
Discovery of SARS-CoV-2 main protease inhibitors using a synthesis-directed de novo design model
/
Aaron Morris, William McCorkindale, the COVID Moonshot Consortium, Nir Drayman, John D Chodera, Savaş Tay, Nir London, and Alpha A. Lee.
Chemical Communications 57:5909, 2021
[DOI]
We show how a machine learning models of ligand affinity can be coupled to synthetic enumeration models to rapidly generate potent inhibitors of the SARS-CoV-2 main viral protease.
What Markov State Models can and cannot do: Correlation versus path-based observables in protein-folding models
/
Ernesto Suárez, Rafal P Wiewiora, Chris Wehmeyer, Frank Noé, John D Chodera, Daniel M Zuckerman
Journal of Chemical Theory and Computation 17:3119, 2021
[DOI] [PDF] [bioRxiv] [GitHub]
Markov state models are now well-established for describing the long-time conformational dynamics of proteins. Here, we take a critical look of what properties can reliably be extracted from these coarse-grained models.
Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity
/
Emma C. Thompson, Laura E. Rosen, James G. Shepherd, Robert Spreafico, Ana da Silva Filipe, Jason A. Wojcechowskyj, Chris Davis, Luca Piccoli, David J. Pascall, Josh Dillen, Spyros Lytras, Nadine Czudnochowski, Rajiv Shah, Marcel Meury, Natasha Jesudason, Anna De Marco, Kathy Li, Jessia Bassi, Aine O’Toole, Dora Pinto, Rachel M. Colqohoun, Katja Culap, Ben Jackson, Fabrizia Zatta, Andrew Rambaut, Stefano Jaconi, Vattipali B. Sreenu, Jay Nix, Ivy Zhang, Ruth F. Jarrett, William G. Glass, Martina Beltramello, Kyriaki Nomikou, Matteo Pizzuto, Lily Tong, Elisabetta Cameroni, Tristan I. Croll, Natasha Johnson, Julia Di Iulio, Arthur Wickenhagen, Alessandro Ceschi, Aoife M. Harbison, Daniel Mair, Paolo Ferrari, Katherine Smollett, Federica Sallusto, Stephen Carmichael, Christian Garzoni, Jenna Nichols, Massimo Galli, Joseph Hughes, Agostino Riva, Antonia Ho, Marco Schiuma, Malcolm G. Semple, Peter J. M. Openshaw, Elisa Fadda, J. Kenneth Baillie, John D. Chodera, The ISARIC4C Investigators, the COVID-19 Genomics UK (COG-UK) consortium, Suzannah J. Rihn, Samantha J. Lycett, Herbert W. Virgin, Amalio Telenti, Davide Corti, David L. Robertson, and Gyorgy Snell.
Cell 184:1171, 2022. [DOI] [PDF] [bioRxiv] [Supplementary Info] [Folding@home data]
New mutations that enhance the affinity of SARS-CoV-2 spike protein for human ACE2—and potentially pose threats to antibody-based therapeutics and vaccines for COVID-19—are already emerging in the wild. We characterize and describe sentinel mutations of SARS-CoV-2 in the wild that herald challenges for combatting COVID-19, and use simulations of the RBD-ACE2 interface on Folding@home to biophysically characterize why these mutations can lead to enhanced affinity.
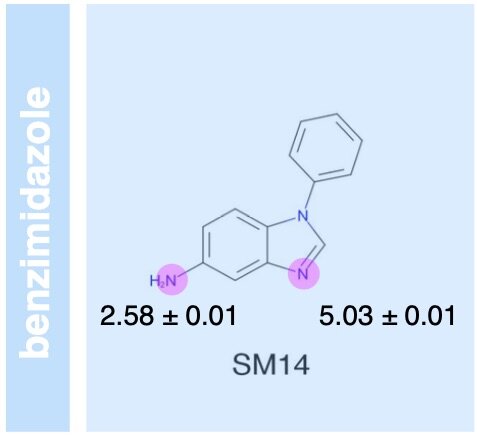
Overview of the SAMPL6 pKa challenge: evaluating small molecule microscopic and macroscopic pKa predictions
/
Mehtap Işık, Ariën S Rustenburg, Andrea Rizzi, Marilyn R Gunner, David L Mobley, John D Chodera
Journal of Computer-Aided Molecular Design 35:131, 2021
[DOI] [bioRxiv] [GitHub] [manuscript and figure sources]
The SAMPL6 pKa challenge assessed the ability of the computational chemistry community to predict macroscopic and microscopic pKas for a set of druglike molecules resembling kinase inhibitors. This paper reports on the overall performance and lessons learned, including the surprising finding that many tools predict reasonably accurate macroscopic pKas corresponding to the wrong microscopic protonation sites.
Crowdsourcing drug discovery for pandemics
/John D. Chodera, Alpha A. Lee, Nir London, and Frank von Delft.
Nature Chemistry 12:581, 2020
[DOI] [PDF] [COVID Moonshot] [GoFundMe]
The COVID-19 pandemic has left the world scrambling to find effective therapies to stem the tidal wave of death and put an end to the worldwide disruption caused by SARS-CoV-2. In this Correspondence, we argue for the need for a new open, collaborative drug discovery model (exemplified by our COVID Moonshot collaboration) that breaks free of the limitations of industry-led competitive drug discovery efforts that necessarily restrict information flow and hinder rapid progress by prioritizing profits and patent protection over human lives.
Standard state free energies, not pKas, are ideal for describing small molecule protonation and tautomeric states
/
M R Gunner, Taichi Murakami, Ariën S. Rustenburg, Mehtap Işık, and John D. Chodera.
Journal of Computer Aided Molecular Design 34:561, 2020. [DOI] [PDF] [GitHub]
Here, we demonstrate how the physical nature of protonation and tautomeric state effects means that the standard state free energies of each microscopic protonation/tautomeric state at a single pH is sufficient to describe the complete pH-dependent microscopic and macroscopic populations. We introduce a new kind of diagram that uses this concept to illustrate a variety of pH-dependent phenomena, and show how it can be used to identify common issues with protonation state prediction algorithms. As a result, we recommend future blind prediction challenges utilize microstate free energies at a single reference pH as the minimal sufficient information for assessing prediction accuracy and utility.
Assessing the accuracy of octanol-water partition coefficient predictions in the SAMPL6 Part II log P Challenge
/
Mehtap Işık, Teresa Danielle Bergazin, Thomas Fox, Andrea Rizzi, John D. Chodera, and David L. Mobley.
Journal of Computer Aided Molecular Design, 34:335, 2020. [DOI] [PDF] [bioRxiv] [GitHub]
We report the performance assessment of the 91 methods that were submitted to the SAMPL6 blind challenge for predicting octanol-water partition coefficient (logP) measurements. The average RMSE of the most accurate five MM-based, QM-based, empirical, and mixed approach methods based on RMSE were 0.92±0.13, 0.48±0.06, 0.47±0.05, and 0.50±0.06, respectively.