
SEARCH
ARTICLE TYPES

Machine-learned molecular mechanics force field for the simulation of protein-ligand systems and beyond
/
Takaba K, Pulido I, Behara PK, Henry M, MacDermott-Opeskin H, Chodera JD, Wang Y
preprint: [arXiv]
We present a new self-consistent MM force field trained on $>$1.1M quantum chemical calculations that uses graph nets to achieve high accuracy and produce accurate protein-ligand binding free energies.
Benchmarking cross-docking strategies for structure-informed machine learning in kinase drug discovery
/
Schaller D, Christ CD, Chodera JD, Volkamer A
preprint: [bioRxiv]
We assess strategies for predicting useful docked ligand poses for structure-informed machine learning for kinase inhibitor drug discovery.
NNP/MM: Fast molecular dynamics simulations with machine learning potentials and molecular mechanics
/
Galvelis R, Varela-Rial A, Doerr S, Fino R, Eastman P, Markland TE, Chodera JD, and de Fabritiis G
Journal of Chemical Information and Modeling 63:5701, 2023 [DOI] [arXiv]
We demonstrate that a new generation of quantum machine learning (QML) potentials based on neural networks---which can achieve quantum chemical accuracy at a fraction of the cost---can be implemented efficiently in the OpenMM molecular dynamics simulation engine as part of hybrid machine learning / molecular mechanics (ML/MM) potentials that promise to deliver superior accuracy for modeling protein-ligand interactions.
Development and benchmarking of Open Force Field 2.0.0---the Sage small molecule force field
/
Boothroyd S, Behara PK, Madin OC, Hahn DF, Jang H, Gapsys V, Wagner JR, Horton JT, Dotson DL, Thompson MW, Maat J, Gokey T, Wang L-P, Cole DJ, Gilson MK, Chodera JD, Bayly CI, Shirts MR, Mobley DL
Journal of Chemical Theory and Computation 19:3251, 2023 [DOI] [chemRxiv] [GitHub] [examples]
We present a new generation of small molecule force field for molecular design from the Open Force Field Initiative fit to both quantum chemical and experimental liquid mixture data
EspalomaCharge: Machine learning-enabled ultra-fast partial charge assignment
/
Wang Y, Pulido I, Takaba K, Kaminow B, Scheen J, Wang L, Chodera JD
preprint: [arXiv]
We present a drop-in replacement for generating AM1-BCC ELF10 charges based on graph convolutional nets that is orders of magnitude faster than standard methods for both small molecules and biomolecules.
Spatial attention kinetic network with E(n) equivariance
/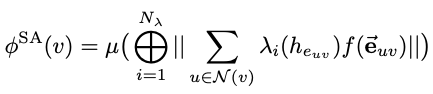
Yuanqing Wang and John D. Chodera
preprint: [arXiv] [code]
This work descibes Spatial Attention Kinetic Networks (SAKE), a new E(n)-equivariant architecture that uses spatial attention, enabling the construction of extremely performant but still accurate machine learning potentials, as well as flows capable of prediction dynamics.
Open Force Field BespokeFit: Automating Bespoke Torsion Parametrization at Scale
/
Horton JT, Boothroyd S, Wagner W, Mitchell JA, Gokey T, Dotson DL, Behara PK, Ramaswamy VK, Mackey M, Chodera JD, Anwar J, Mobley DL, and Cole DJ
Journal of Chemical Informatics and Modeling 62:22, 2022 [DOI]
We describe an automated pipeline for generating tailored force field parameters for small molecules using quantum chemical or quantum machine learning potentials.
End-to-end differentiable molecular mechanics force field construction
/
Yuanqing Wang, Josh Fass, and John D. Chodera
Chemical Science 13:12016, 2022 [DOI] [arXiv] [pytorch code] [JAX code]
Molecular mechanics force fields have been a workhorse for computational chemistry and drug discovery. Here, we propose a new approach to force field parameterization in which graph convolutional networks are used to perceive chemical environments and assign molecular mechanics (MM) force field parameters. The entire process of chemical perception and parameter assignment is differentiable end-to-end with respect to model parameters, allowing new force fields to be easily constructed from MM or QM force fields, extended, and applied to arbitrary biomolecules.
Capturing non-local through-bond effects in molecular mechanics force fields: II. Using fractional bond orders to fit torsion parameters
/
Stern CD, Maat J, Dotson DL, Bayly CI, Smith DGA, Mobley DL, and Chodera JD
preprint: [bioRxiv]
We show how the Wiberg Bond Order (WBO) can be used to accurately interpolate torsional profiles for molecular mechanics force fields, which holts the potential for drastically reducing the complexity of these force fields while increasing their ability to generalize and accurately treat complex druglike molecules such as kinase inhibitors.
GCN2 kinase activation by ATP-competitive kinase inhibitors
/
Mellinghoff I, Tang CP, Clark O, Ferrarone J, Campos C, Lalani AS, Chodera JD, Intlekofer AM, and Elemento O
Nature Chemical Biology}, 18:207, 2022 [DOI]
We describe paradoxical activation of GCN2 kinase activity by the kinase inhibitor neratinib, and propose a model for how inhibitor-induced dimerization might cause this unusual activity.
Teaching free energy calculations to learn from experimental data
/
Marcus Wieder, Josh Fass, and John Chodera
[bioRxiv] [code] [data]
We show, for the first time, how alchemical free energy calculations can be used to not only compute free energy differences between small molecules involving covalent bond rearrangements in systems treated entirely with quantum machine learning potentials, but that these calculations have the capacity to learn to efficiently generalize from conditioning on experimental free energy data.
The Open Force Field Evaluator: An automated, efficient, and scalable framework for the estimation of physical properties from molecular simulation
/
Simon Boothroyd, Lee-Ping Wang, David L. Mobley, John D. Chodera, and Michael R. Shirts
Preprint ahead of submission: [ChemRxiv]
We describe a new software framework for automated evaluation of physical properties for the benchmarking and optimization of small molecule force fields according to best practices.
Antibodies to the SARS-CoV-2 receptor-binding domain that maximize breadth and resistance to viral escape
/
Tyler N Starr, Nadine Czudnochowski, Fabrizia Zatta, Young-Jun Park, Zhuoming Liu, Amin Addetia, Dora Pinto, Martina Beltramello, Patrick Hernandez, Allison J Greaney, Roberta Marzi, William G Glass, Ivy Zhang, Adam S Dingens, John E Bowen, Jason A Wojcechowskyj, Anna De Marco, Laura E Rosen, Jiayi Zhou, Martin Montiel-Ruiz, Hannah Kaiser, Heather Tucker, Michael P Housley, Julia Di Iulio, Gloria Lombardo, Maria Agostini, Nicole Sprugasci, Katja Culap, Stefano Jaconi, Marcel Meury, Exequiel Dellota, Elisabetta Cameroni, Tristan I Croll, Jay C Nix, Colin Havenar-Daughton, Amalio Telenti, Florian A Lempp, Matteo Samuele Pizzuto, John D Chodera, Christy M Hebner, Sean PJ Whelan, Herbert W Virgin, David Veesler, Davide Corti, Jesse D Bloom, Gyorgy Snell
Nature, in press. [DOI] [bioRxiv] [GitHub]
We comprehensively characterize escape, breadth, and potency across a panel of SARS-CoV-2 antibodies targeting the receptor binding domain, including the parent antibody of the recently approved Vir antibody drug (Sotrovimab), illuminating escape mutations with structural and dynamic insight into their mechanism of action.
Mutation in Abl kinase with altered drug binding kinetics indicates a novel mechanism of imatinib resistance
/
Agatha Lyczek, Benedict Tilman Berger, Aziz M Rangwala, YiTing Paung, Jessica Tom, Hannah Philipose, Jiaye Guo, Steven K Albanese, Matthew B Robers, Stefan Knapp, John D Chodera, Markus A Seeliger
Preprint ahead of publication: [bioRxiv]
Here, we characterize the biophysical mechanisms underlying mutants of Abl kinase associated with clinical drug resistance to targeted cancer therapies. We uncover a surprising novel mechanism of mutational resistance to kinase inhibitor therapy in which the off-rate for inhibitor unbinding is increased without affecting inhibitor affinity.
Best practices for constructing, preparing, and evaluating protein-ligand binding affinity benchmarks
/
David F Hahn, Christopher I Bayly, Hannah E Bruce Macdonald, John D Chodera, Antonia SJS Mey, David L Mobley, Laura Perez Benito, Christina EM Schindler, Gary Tresadern, Gregory L Warren
Preprint ahead of publication: [arXiv] [GitHub]
This living best practices paper for the Living Journal of Computational Molecular Sciences describes the current community consensus in how to curate experimental benchmark data for assessing predictive affinity models for drug discovery, how to prepare these systems for affinity calculations, and how to assess the results to compare performance.
Bayesian inference-driven model parameterization and model selection for 2CLJQ fluid models
/
Owen C Madin, Simon Boothroyd, Richard A Messerly, John D Chodera, Josh Fass, and Michael R Shirts
Preprint ahead of publication: [arXiv]
Here, we show how Bayesian inference can be used to automatically perform model selection and fit parameters for a molecular mechanics force field.
Fitting quantum machine learning potentials to experimental free energy data: Predicting tautomer ratios in solution
/
Marcus Wieder, Josh Fass, and John D. Chodera
Chemical Science, in press [bioRxiv] [code]
We demonstrate, for the first time, how alchemical free energy calculations can performed on systems simulated entirely with quantum machine learning potentials and how these potentials can be retrained on experimental free energies to generalize to new molecules from limited training data. We apply this approach to a difficult problem in small molecule drug discovery: Predicting accurate tautomer ratios in solution.
Capturing non-local through-bond effects when fragmenting molecules for quantum chemical torsion scans
/
Chaya D. Stern, Christopher I. Bayly, Daniel G. A. Smith, Josh Fass, Lee-Ping Wang, David L. Mobley, and John D. Chodera
Preprint ahead of submission
[bioRxiv] [GitHub code] [GitHub data] [Metadata] [OSF]
Best practices for alchemical free energy calculations
/
Mey ASJS, Allen B, Bruce Macdonald HE, Chodera JD, Kuhn M, Michel J, Mobley DL, Naden LN, Prasad S, Rizzi A, Scheen J, Shirts MR, Tresadern G, and Xu H.
Living Journal of Computational Molecular Sciences 2022 [DOI]
[arXiv] [GitHub]
This living review for the Living Journal of Computational Molecular Sciences (LiveCoMS) covers the essential considerations for running alchemical free energy calculations for rational molecular design for drug discovery.