
SEARCH
ARTICLE TYPES

Capturing non-local through-bond effects in molecular mechanics force fields: II. Using fractional bond orders to fit torsion parameters
/
Stern CD, Maat J, Dotson DL, Bayly CI, Smith DGA, Mobley DL, and Chodera JD
preprint: [bioRxiv]
We show how the Wiberg Bond Order (WBO) can be used to accurately interpolate torsional profiles for molecular mechanics force fields, which holts the potential for drastically reducing the complexity of these force fields while increasing their ability to generalize and accurately treat complex druglike molecules such as kinase inhibitors.
Teaching free energy calculations to learn from experimental data
/
Marcus Wieder, Josh Fass, and John Chodera
[bioRxiv] [code] [data]
We show, for the first time, how alchemical free energy calculations can be used to not only compute free energy differences between small molecules involving covalent bond rearrangements in systems treated entirely with quantum machine learning potentials, but that these calculations have the capacity to learn to efficiently generalize from conditioning on experimental free energy data.
Antibodies to the SARS-CoV-2 receptor-binding domain that maximize breadth and resistance to viral escape
/
Tyler N Starr, Nadine Czudnochowski, Fabrizia Zatta, Young-Jun Park, Zhuoming Liu, Amin Addetia, Dora Pinto, Martina Beltramello, Patrick Hernandez, Allison J Greaney, Roberta Marzi, William G Glass, Ivy Zhang, Adam S Dingens, John E Bowen, Jason A Wojcechowskyj, Anna De Marco, Laura E Rosen, Jiayi Zhou, Martin Montiel-Ruiz, Hannah Kaiser, Heather Tucker, Michael P Housley, Julia Di Iulio, Gloria Lombardo, Maria Agostini, Nicole Sprugasci, Katja Culap, Stefano Jaconi, Marcel Meury, Exequiel Dellota, Elisabetta Cameroni, Tristan I Croll, Jay C Nix, Colin Havenar-Daughton, Amalio Telenti, Florian A Lempp, Matteo Samuele Pizzuto, John D Chodera, Christy M Hebner, Sean PJ Whelan, Herbert W Virgin, David Veesler, Davide Corti, Jesse D Bloom, Gyorgy Snell
Nature, in press. [DOI] [bioRxiv] [GitHub]
We comprehensively characterize escape, breadth, and potency across a panel of SARS-CoV-2 antibodies targeting the receptor binding domain, including the parent antibody of the recently approved Vir antibody drug (Sotrovimab), illuminating escape mutations with structural and dynamic insight into their mechanism of action.
Mutation in Abl kinase with altered drug binding kinetics indicates a novel mechanism of imatinib resistance
/
Agatha Lyczek, Benedict Tilman Berger, Aziz M Rangwala, YiTing Paung, Jessica Tom, Hannah Philipose, Jiaye Guo, Steven K Albanese, Matthew B Robers, Stefan Knapp, John D Chodera, Markus A Seeliger
Preprint ahead of publication: [bioRxiv]
Here, we characterize the biophysical mechanisms underlying mutants of Abl kinase associated with clinical drug resistance to targeted cancer therapies. We uncover a surprising novel mechanism of mutational resistance to kinase inhibitor therapy in which the off-rate for inhibitor unbinding is increased without affecting inhibitor affinity.
What Markov State Models can and cannot do: Correlation versus path-based observables in protein-folding models
/
Ernesto Suárez, Rafal P Wiewiora, Chris Wehmeyer, Frank Noé, John D Chodera, Daniel M Zuckerman
Journal of Chemical Theory and Computation 17:3119, 2021
[DOI] [PDF] [bioRxiv] [GitHub]
Markov state models are now well-established for describing the long-time conformational dynamics of proteins. Here, we take a critical look of what properties can reliably be extracted from these coarse-grained models.
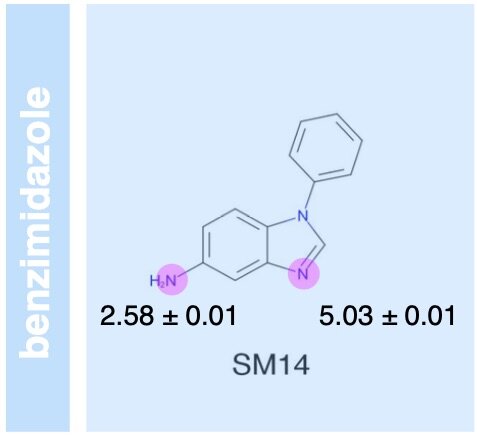
Overview of the SAMPL6 pKa challenge: evaluating small molecule microscopic and macroscopic pKa predictions
/
Mehtap Işık, Ariën S Rustenburg, Andrea Rizzi, Marilyn R Gunner, David L Mobley, John D Chodera
Journal of Computer-Aided Molecular Design 35:131, 2021
[DOI] [bioRxiv] [GitHub] [manuscript and figure sources]
The SAMPL6 pKa challenge assessed the ability of the computational chemistry community to predict macroscopic and microscopic pKas for a set of druglike molecules resembling kinase inhibitors. This paper reports on the overall performance and lessons learned, including the surprising finding that many tools predict reasonably accurate macroscopic pKas corresponding to the wrong microscopic protonation sites.
Capturing non-local through-bond effects when fragmenting molecules for quantum chemical torsion scans
/
Chaya D. Stern, Christopher I. Bayly, Daniel G. A. Smith, Josh Fass, Lee-Ping Wang, David L. Mobley, and John D. Chodera
Preprint ahead of submission
[bioRxiv] [GitHub code] [GitHub data] [Metadata] [OSF]
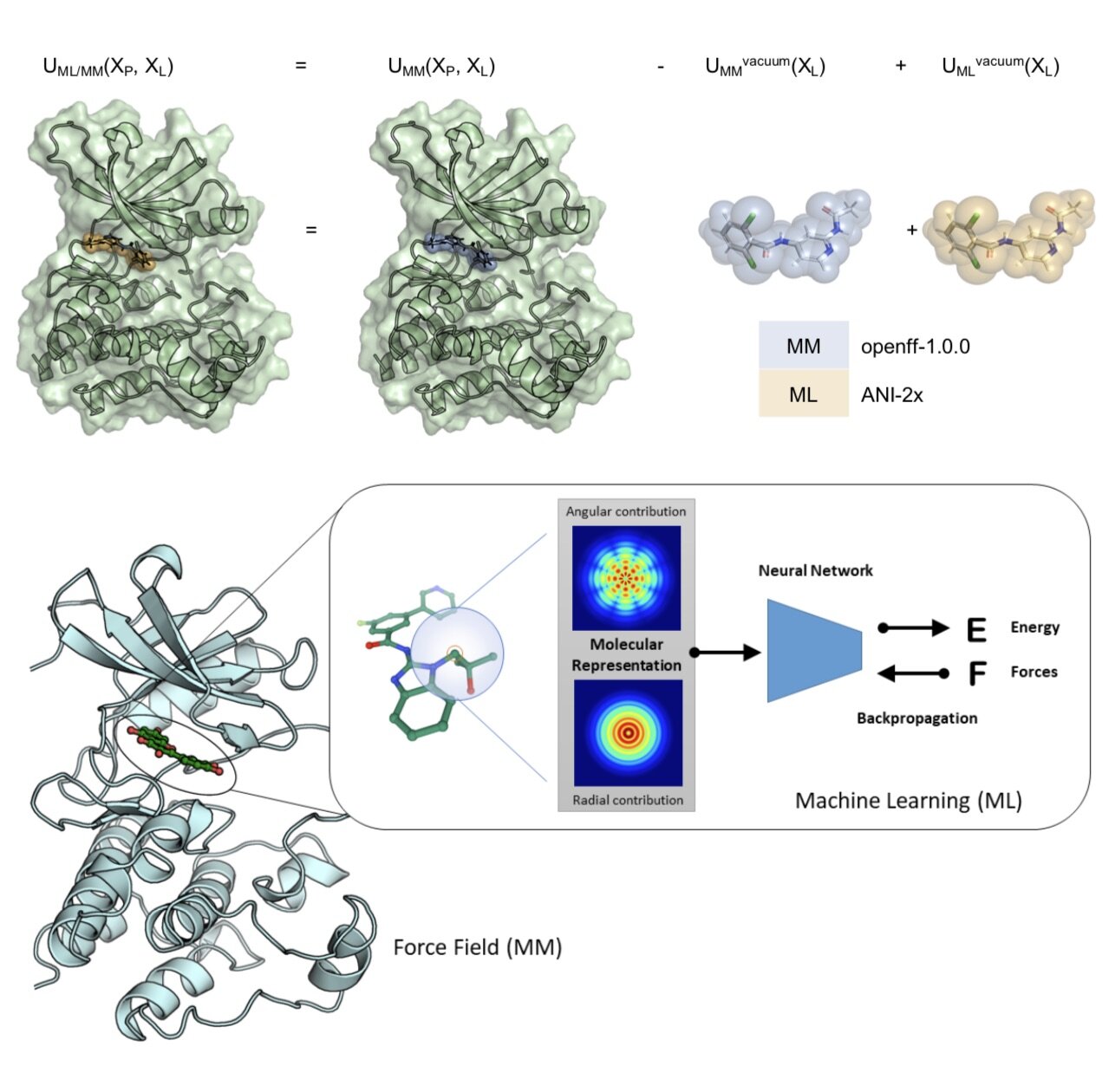
Towards chemical accuracy for alchemical free energy calculations with hybrid physics-based machine learning / molecular mechanics potentials
/
Dominic A. Rufa, Hannah E. Bruce Macdonald, Josh Fass, Marcus Wieder, Patrick B. Grinaway, Adrian E. Roitberg, Olexandr Isayev, and John D. Chodera.
Preprint ahead of submission.
[bioRxiv] [GitHub]
In this first use of hybrid machine learning / molecular mechanics (ML/MM) potentials for alchemical free energy calculations, we demonstrate how the improved modeling of intramolecular ligand energetics offered by the quantum machine learning potential ANI-2x can significantly improve the accuracy in predicting kinase inhibitor binding free energy by reducing the error from 0.97~kcal/mol to 0.47~kcal/mol, which could drastically reduce the number of compounds that must be synthesized in lead optimization campaigns for minimal additional computational cost.
Is structure based drug design ready for selectivity optimization?
/
Steven K. Albanese, John D. Chodera, Andrea Volkamer, Simon Keng, Robert Abel, and Lingle Wang
Journal of Chemical Informatics and Modeling [DOI] [bioRxiv] [GitHub]
We asked whether the similarity of binding sites in related kinases might result in a fortuitous cancellation of errors in using alchemical free energy calculations to predict kinase inhibitor selectivities. Surprisingly, we find that even distantly related kinases have sufficient correlation in their errors that predicting changes in selectivity can be much more accurate than predicting changes in potency due to this effect, and show how this could lead to large reductions in the number of molecules that must be synthesized to achieve a desired selectivity goal.
The SAMPL6 SAMPLing challenge: Assessing the reliability and efficiency of binding free energy calculations
/
Andrea Rizzi, Travis Jensen, David R. Slochower, Matteo Aldeghi, Vytautas Gapsys, Dimitris Ntekoumes, Stefano Bosisio, Michail Papadourakis, Niel M. Henriksen, Bert L. de Groot, Zoe Cournia, Alex Dickson, Julien Michel, Michael K. Gilson, Michael R. Shirts, David L. Mobley, and John D. Chodera
Journal of Computer Aided Molecular Design 34:601, 2020. [DOI] [PDF] [bioRxiv] [GitHub]
To assess the relative efficiencies of alchemical binding free energy calculations, the SAMPL6 SAMPLing challenge asked participants to submit predictions as a function of computer effort for the same force field and charge model. Surprisingly, we found that most molecular simulation codes cannot agree on the binding free energy was, even for the same force field.
Octanol-water partition coefficient measurements for the SAMPL6 Blind Prediction Challenge
/
Mehtap Işık, Dorothy Levorse, David L. Mobley, Timothy Rhodes, and John D. Chodera.
Journal of Computer Aided Molecular Design 34:405, 2020. [DOI] [bioRxiv] [data] [GitHub]
We describe the design and data collection (and associated challenges) for the SAMPL6 part II logP octanol-water blind prediction challenge, where the goal was to benchmark the accuracy of force fields for druglike molecules (here, molecules resembling kinase inhibitors).
OpenPathSampling: A Python framework for path sampling simulations. II. Building and customizing path ensembles and sample schemes
/David W.H. Swenson, Jan-Hendrik Prinz, Frank Noé, John D. Chodera, Peter G. Bolhuis
Journal of Chemical Theory and Computation 15:837, 2019. [DOI] [bioRxiv] [PDF] [GitHub] [openpathsampling.org]
To make powerful path sampling techniques broadly accessible and efficient, we have produced a new Python framework for easily implementing path sampling strategies (such as transition path and interface sampling) in Python. This second publication describes advanced aspects of the theory and details of how to customize path ensembles.
OpenPathSampling: A Python framework for path sampling simulations. I. Basics
/David W.H. Swenson, Jan-Hendrik Prinz, Frank Noé, John D. Chodera, Peter G. Bolhuis
Journal of Chemical Theory and Computation 15:813, 2019 [DOI] [bioRxiv] [PDF] [GitHub] [openpathsampling.org]
To make powerful path sampling techniques broadly accessible and efficient, we have produced a new Python framework for easily implementing path sampling strategies (such as transition path and interface sampling) in Python. This first publication describes some of the theory and capabilities behind the approach.
Overview of the SAMPL6 host-guest binding affinity prediction challenge
/
Andrea Rizzi, Steven Murkli, John N. McNeill, Wei Yao, Matthew Sullivan, Michael K. Gilson, Michael W. Chiu, Lyle Isaacs, Bruce C. Gibb, David L. Mobley*, John D. Chodera*
* denotes co-corresponding authors
Journal of Computer-Aided Molecular Design special issue on SAMPL6, 32:937, 2018. [DOI] [bioRxiv] [GitHub]
We present an overview of the host-guest systems and participant performance for the SAMPL6 host-guest blind affinity prediction challenges, assessing how well various physical modeling approaches were able to predict ligand binding affinities for simple ligand recognition problems where receptor sampling and protonation state effects are eliminated due to the simplicity of supramolecular hosts. We find that progress is now stagnated likely due to force field limitations.
Biomolecular simulations under realistic macroscopic salt conditions
/
Gregory A. Ross, Ariën S. Rustenburg, Patrick B. Grinaway, Josh Fass, and John D. Chodera
Journal of Physical Chemistry B 122:5466, 2018. [DOI] [bioRxiv] [simulation code] [results and analysis scripts]
We show how NCMC can be used to implement an efficient osmostat in molecular dynamics simulations to model realistic fluctuations in ion environments around biomolecules, and illustrate how the local salt environment around biological macromolecules can differ substantially from bulk.
OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics
/
Peter Eastman, Jason Swails, John D. Chodera, Robert T. McGibbon, Yutong Zhao, Kyle A. Beauchamp, Lee-Ping Wang, Andrew C. Simmonett, Matthew P. Harrigan, Chaya D. Stern, Rafal P. Wiewiora, Bernard R. Brooks, Vijay S. Pande. PLoS Computational Biology 13:e1005659, 2017. [DOI] [bioRxiv] [website] [GitHub]
We describe the latest version of OpenMM, a GPU-accelerated framework for high performance molecular simulation applications.
Approaches for calculating solvation free energies and enthalpies demonstrated with an update of the FreeSolv database
/
Guilherme Duarte Ramos Matos, Daisy Y. Kyu, Hannes H. Loeffler, John D. Chodera, Michael R. Shirts, David Mobley
Journal of Chemical Engineering Data 62:1559, 2017. [DOI] [bioRxiv] [GitHub]
We review alchemical methods for computing solvation free energies and present an update (version 0.5) to the FreeSolv database of experimental and calculated hydration free energies of neutral compounds.