
SEARCH
ARTICLE TYPES

Open science discovery of potent noncovalent SARS-CoV-2 main protease inhibitors
/Boby ML, Fearon D, Ferla M, Filep M, Robinson MC, The COVID Moonshot Consortium, Chodera JD, Lee A, London N, von Delft F.
Science 382:eabo7201, 2023 [DOI] [ready to use data]
We report the discovery of a new oral antiviral non-covalent SARS-CoV-2 main protease inhibitor developed by the COVID Moonshot, a global open science collaboration leveraging free energy calculations on Folding@home and ML-accelerated synthesis planning, now in accelerated preclinical studies funded by an $11M grant from the WHO ACT-A program via the Wellcome Trust. We are currently in discussions with generics manufacturers about partnering with us throughout clinical trials to ensure we can scale up production for global equitable and affordable access once approved by regulatory agencies.
Benchmarking cross-docking strategies for structure-informed machine learning in kinase drug discovery
/
Schaller D, Christ CD, Chodera JD, Volkamer A
preprint: [bioRxiv]
We assess strategies for predicting useful docked ligand poses for structure-informed machine learning for kinase inhibitor drug discovery.
NNP/MM: Fast molecular dynamics simulations with machine learning potentials and molecular mechanics
/
Galvelis R, Varela-Rial A, Doerr S, Fino R, Eastman P, Markland TE, Chodera JD, and de Fabritiis G
Journal of Chemical Information and Modeling 63:5701, 2023 [DOI] [arXiv]
We demonstrate that a new generation of quantum machine learning (QML) potentials based on neural networks---which can achieve quantum chemical accuracy at a fraction of the cost---can be implemented efficiently in the OpenMM molecular dynamics simulation engine as part of hybrid machine learning / molecular mechanics (ML/MM) potentials that promise to deliver superior accuracy for modeling protein-ligand interactions.
Development and benchmarking of Open Force Field 2.0.0---the Sage small molecule force field
/
Boothroyd S, Behara PK, Madin OC, Hahn DF, Jang H, Gapsys V, Wagner JR, Horton JT, Dotson DL, Thompson MW, Maat J, Gokey T, Wang L-P, Cole DJ, Gilson MK, Chodera JD, Bayly CI, Shirts MR, Mobley DL
Journal of Chemical Theory and Computation 19:3251, 2023 [DOI] [chemRxiv] [GitHub] [examples]
We present a new generation of small molecule force field for molecular design from the Open Force Field Initiative fit to both quantum chemical and experimental liquid mixture data
Spatial attention kinetic network with E(n) equivariance
/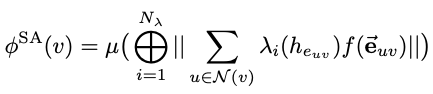
Yuanqing Wang and John D. Chodera
preprint: [arXiv] [code]
This work descibes Spatial Attention Kinetic Networks (SAKE), a new E(n)-equivariant architecture that uses spatial attention, enabling the construction of extremely performant but still accurate machine learning potentials, as well as flows capable of prediction dynamics.
Open Force Field BespokeFit: Automating Bespoke Torsion Parametrization at Scale
/
Horton JT, Boothroyd S, Wagner W, Mitchell JA, Gokey T, Dotson DL, Behara PK, Ramaswamy VK, Mackey M, Chodera JD, Anwar J, Mobley DL, and Cole DJ
Journal of Chemical Informatics and Modeling 62:22, 2022 [DOI]
We describe an automated pipeline for generating tailored force field parameters for small molecules using quantum chemical or quantum machine learning potentials.
End-to-end differentiable molecular mechanics force field construction
/
Yuanqing Wang, Josh Fass, and John D. Chodera
Chemical Science 13:12016, 2022 [DOI] [arXiv] [pytorch code] [JAX code]
Molecular mechanics force fields have been a workhorse for computational chemistry and drug discovery. Here, we propose a new approach to force field parameterization in which graph convolutional networks are used to perceive chemical environments and assign molecular mechanics (MM) force field parameters. The entire process of chemical perception and parameter assignment is differentiable end-to-end with respect to model parameters, allowing new force fields to be easily constructed from MM or QM force fields, extended, and applied to arbitrary biomolecules.
Improving force field accuracy by training against condensed-phase mixture properties
/
Boothroyd S, Madin OC, Mobley DL, Wang L-P, Chodera JD, and Shirts MR
Journal of Chemical Theory and Computation 18:3577, 2022 [DOI] [GitHub]
We use a new automated framework for physical property evaluation and fitting to show how molecular mechanics force fields can be systematically improved by fitting to condensed phase properties.
Capturing non-local through-bond effects in molecular mechanics force fields: II. Using fractional bond orders to fit torsion parameters
/
Stern CD, Maat J, Dotson DL, Bayly CI, Smith DGA, Mobley DL, and Chodera JD
preprint: [bioRxiv]
We show how the Wiberg Bond Order (WBO) can be used to accurately interpolate torsional profiles for molecular mechanics force fields, which holts the potential for drastically reducing the complexity of these force fields while increasing their ability to generalize and accurately treat complex druglike molecules such as kinase inhibitors.
Teaching free energy calculations to learn from experimental data
/
Marcus Wieder, Josh Fass, and John Chodera
[bioRxiv] [code] [data]
We show, for the first time, how alchemical free energy calculations can be used to not only compute free energy differences between small molecules involving covalent bond rearrangements in systems treated entirely with quantum machine learning potentials, but that these calculations have the capacity to learn to efficiently generalize from conditioning on experimental free energy data.
Mutation in Abl kinase with altered drug binding kinetics indicates a novel mechanism of imatinib resistance
/
Agatha Lyczek, Benedict Tilman Berger, Aziz M Rangwala, YiTing Paung, Jessica Tom, Hannah Philipose, Jiaye Guo, Steven K Albanese, Matthew B Robers, Stefan Knapp, John D Chodera, Markus A Seeliger
Preprint ahead of publication: [bioRxiv]
Here, we characterize the biophysical mechanisms underlying mutants of Abl kinase associated with clinical drug resistance to targeted cancer therapies. We uncover a surprising novel mechanism of mutational resistance to kinase inhibitor therapy in which the off-rate for inhibitor unbinding is increased without affecting inhibitor affinity.
Best practices for constructing, preparing, and evaluating protein-ligand binding affinity benchmarks
/
David F Hahn, Christopher I Bayly, Hannah E Bruce Macdonald, John D Chodera, Antonia SJS Mey, David L Mobley, Laura Perez Benito, Christina EM Schindler, Gary Tresadern, Gregory L Warren
Preprint ahead of publication: [arXiv] [GitHub]
This living best practices paper for the Living Journal of Computational Molecular Sciences describes the current community consensus in how to curate experimental benchmark data for assessing predictive affinity models for drug discovery, how to prepare these systems for affinity calculations, and how to assess the results to compare performance.
Bayesian inference-driven model parameterization and model selection for 2CLJQ fluid models
/
Owen C Madin, Simon Boothroyd, Richard A Messerly, John D Chodera, Josh Fass, and Michael R Shirts
Preprint ahead of publication: [arXiv]
Here, we show how Bayesian inference can be used to automatically perform model selection and fit parameters for a molecular mechanics force field.
What Markov State Models can and cannot do: Correlation versus path-based observables in protein-folding models
/
Ernesto Suárez, Rafal P Wiewiora, Chris Wehmeyer, Frank Noé, John D Chodera, Daniel M Zuckerman
Journal of Chemical Theory and Computation 17:3119, 2021
[DOI] [PDF] [bioRxiv] [GitHub]
Markov state models are now well-established for describing the long-time conformational dynamics of proteins. Here, we take a critical look of what properties can reliably be extracted from these coarse-grained models.
Development and benchmarking of Open Force Field v1.0.0, the Parsley small molecule force field
/
Yudong Qiu, Daniel Smith, Simon Boothroyd, Hyesu Jang, Jeffrey Wagner, Caitlin C Bannan, Trevor Gokey, Victoria T Lim, Chaya Stern, Andrea Rizzi, Xavier Lucas, Bryon Tjanaka, Michael R Shirts, Michael Gilson, John D. Chodera, Christopher I Bayly, David Mobley, Lee-Ping Wang
Preprint ahead of publication: [chemRxiv] [force fields] [Open Force Field Initiative]
We present a new, modern small molecule force field for molecular design from the Open Force Field Initiative, a large industry-academic collaboration that focuses on open science, open data, and modern open source infrastructure.
Capturing non-local through-bond effects when fragmenting molecules for quantum chemical torsion scans
/
Chaya D. Stern, Christopher I. Bayly, Daniel G. A. Smith, Josh Fass, Lee-Ping Wang, David L. Mobley, and John D. Chodera
Preprint ahead of submission
[bioRxiv] [GitHub code] [GitHub data] [Metadata] [OSF]
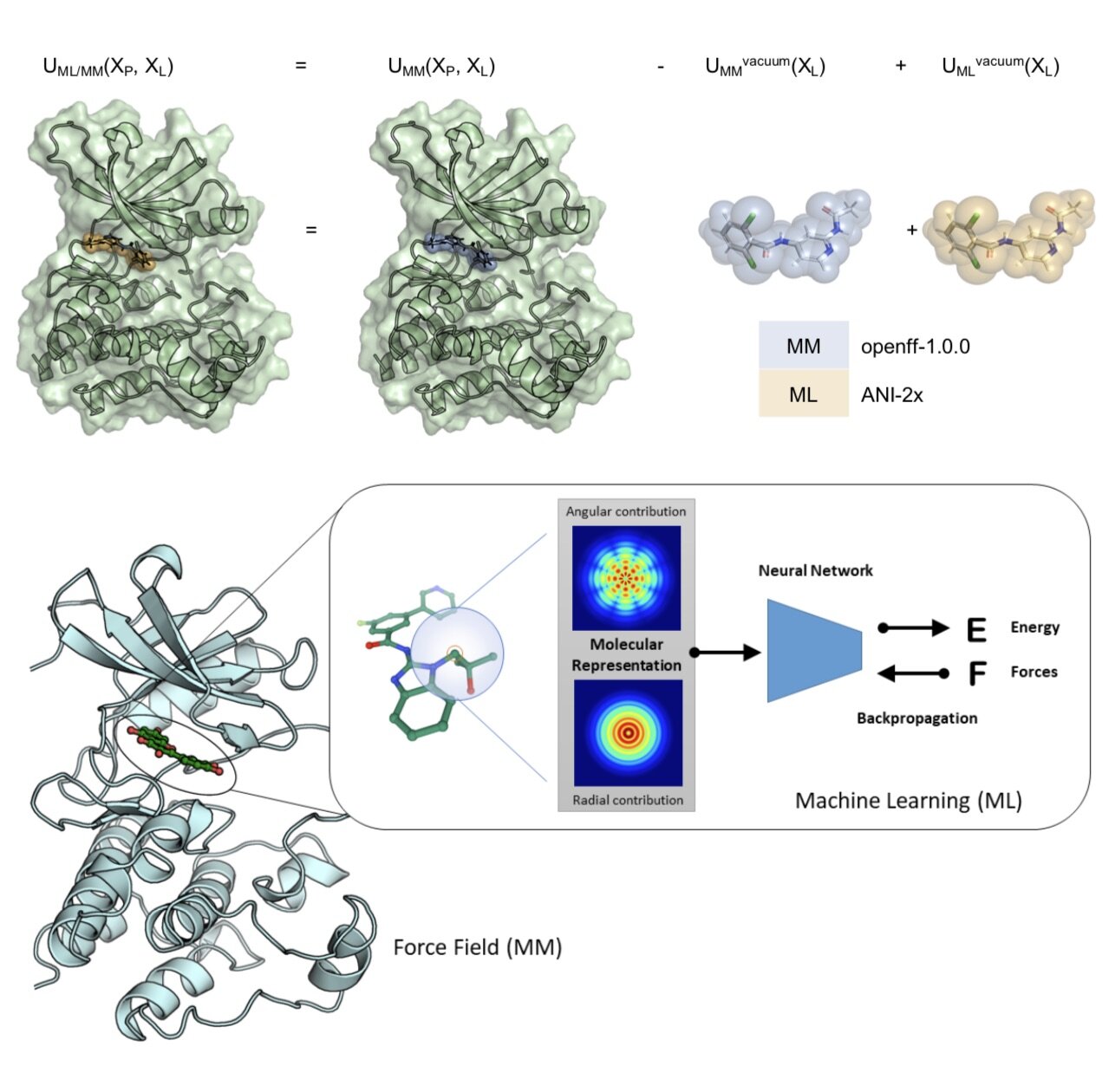
Towards chemical accuracy for alchemical free energy calculations with hybrid physics-based machine learning / molecular mechanics potentials
/
Dominic A. Rufa, Hannah E. Bruce Macdonald, Josh Fass, Marcus Wieder, Patrick B. Grinaway, Adrian E. Roitberg, Olexandr Isayev, and John D. Chodera.
Preprint ahead of submission.
[bioRxiv] [GitHub]
In this first use of hybrid machine learning / molecular mechanics (ML/MM) potentials for alchemical free energy calculations, we demonstrate how the improved modeling of intramolecular ligand energetics offered by the quantum machine learning potential ANI-2x can significantly improve the accuracy in predicting kinase inhibitor binding free energy by reducing the error from 0.97~kcal/mol to 0.47~kcal/mol, which could drastically reduce the number of compounds that must be synthesized in lead optimization campaigns for minimal additional computational cost.
Machine learning force fields and coarse-grained variables in molecular dynamics: application to materials and biological systems
/
Gkeka P, Stoltz G, Farimani AB, Belkacemi Z, Ceriotti M, Chodera JD, Dinner AR, Ferguson A, Maillet JB, Minoux H, Peter C, Pietrucci F, Silveira A, Tkatchenko A, Trstanova Z, Wiewiora R, Leliévre T.
Journal of Chemical Theory and Computation 60:6211, 2020. [DOI] [arXiv]
We review the state of the art in applying machine learning to coarse grain force fields in space and time to study mutliscale dynamics.
The SAMPL6 SAMPLing challenge: Assessing the reliability and efficiency of binding free energy calculations
/
Andrea Rizzi, Travis Jensen, David R. Slochower, Matteo Aldeghi, Vytautas Gapsys, Dimitris Ntekoumes, Stefano Bosisio, Michail Papadourakis, Niel M. Henriksen, Bert L. de Groot, Zoe Cournia, Alex Dickson, Julien Michel, Michael K. Gilson, Michael R. Shirts, David L. Mobley, and John D. Chodera
Journal of Computer Aided Molecular Design 34:601, 2020. [DOI] [PDF] [bioRxiv] [GitHub]
To assess the relative efficiencies of alchemical binding free energy calculations, the SAMPL6 SAMPLing challenge asked participants to submit predictions as a function of computer effort for the same force field and charge model. Surprisingly, we found that most molecular simulation codes cannot agree on the binding free energy was, even for the same force field.