Best practices for constructing, preparing, and evaluating protein-ligand binding affinity benchmarks
/
David F Hahn, Christopher I Bayly, Hannah E Bruce Macdonald, John D Chodera, Antonia SJS Mey, David L Mobley, Laura Perez Benito, Christina EM Schindler, Gary Tresadern, Gregory L Warren
Preprint ahead of publication: [arXiv] [GitHub]
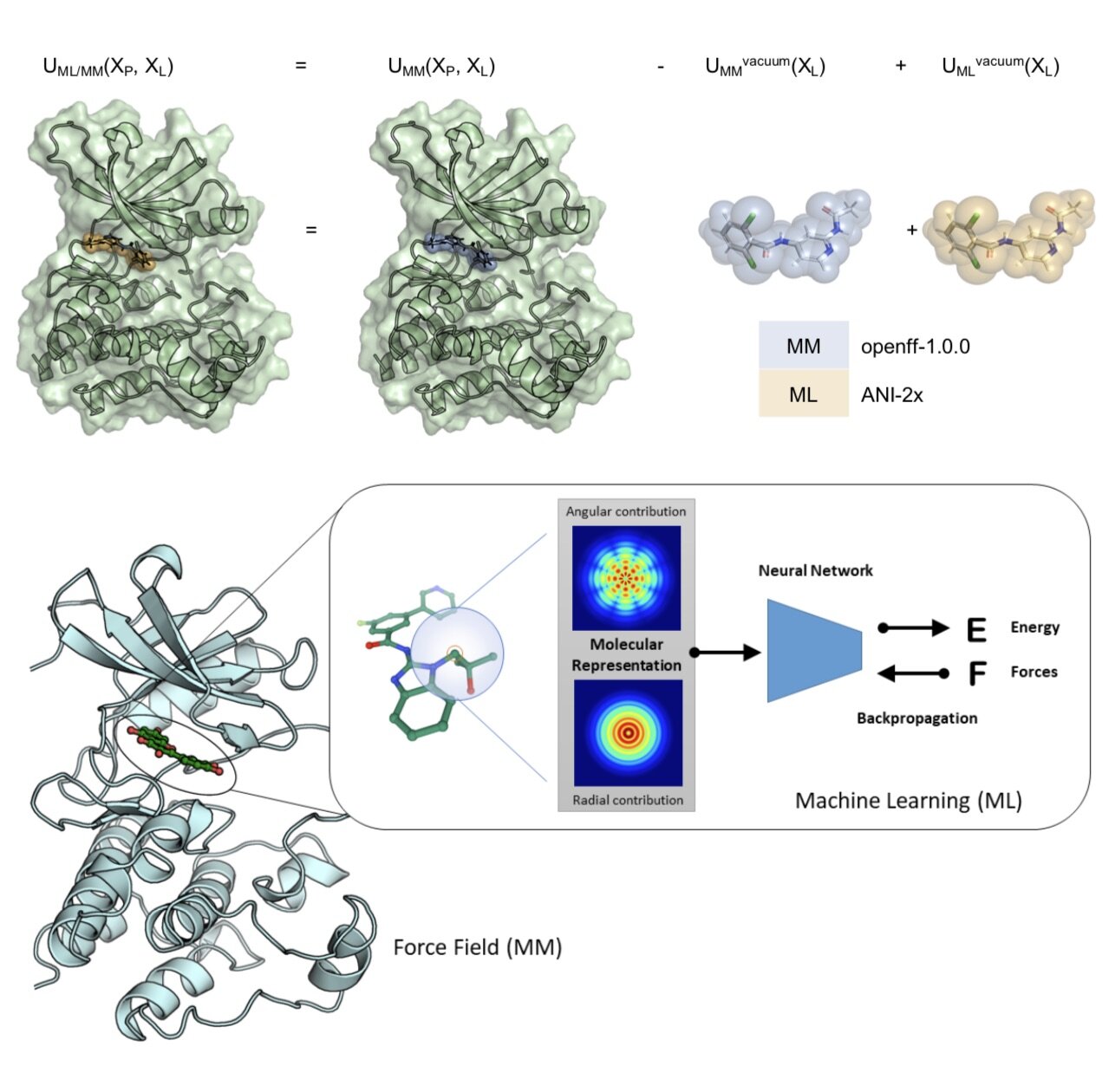
This living best practices paper for the Living Journal of Computational Molecular Sciences describes the current community consensus in how to curate experimental benchmark data for assessing predictive affinity models for drug discovery, how to prepare these systems for affinity calculations, and how to assess the results to compare performance.