
SEARCH
ARTICLE TYPES

Open science discovery of potent noncovalent SARS-CoV-2 main protease inhibitors
/Boby ML, Fearon D, Ferla M, Filep M, Robinson MC, The COVID Moonshot Consortium, Chodera JD, Lee A, London N, von Delft F.
Science 382:eabo7201, 2023 [DOI] [ready to use data]
We report the discovery of a new oral antiviral non-covalent SARS-CoV-2 main protease inhibitor developed by the COVID Moonshot, a global open science collaboration leveraging free energy calculations on Folding@home and ML-accelerated synthesis planning, now in accelerated preclinical studies funded by an $11M grant from the WHO ACT-A program via the Wellcome Trust. We are currently in discussions with generics manufacturers about partnering with us throughout clinical trials to ensure we can scale up production for global equitable and affordable access once approved by regulatory agencies.
NNP/MM: Fast molecular dynamics simulations with machine learning potentials and molecular mechanics
/
Galvelis R, Varela-Rial A, Doerr S, Fino R, Eastman P, Markland TE, Chodera JD, and de Fabritiis G
Journal of Chemical Information and Modeling 63:5701, 2023 [DOI] [arXiv]
We demonstrate that a new generation of quantum machine learning (QML) potentials based on neural networks---which can achieve quantum chemical accuracy at a fraction of the cost---can be implemented efficiently in the OpenMM molecular dynamics simulation engine as part of hybrid machine learning / molecular mechanics (ML/MM) potentials that promise to deliver superior accuracy for modeling protein-ligand interactions.
Identifying and Overcoming the Sampling Challenges in Relative Binding Free Energy Calculations of a Model Protein:Protein Complex
/
Zhang I, Rufa DA, Pulido I, Henry MM, Rosen LE, Hauser K, Singh S, Chodera JD
Journal of Chemical Theory and Computation 19:4863, 2023
We assess what is required for alchemical free energy calculations to be able to make high-quality predictions of the impact of interfacial mutations on protein-protein binding.
Development and benchmarking of Open Force Field 2.0.0---the Sage small molecule force field
/
Boothroyd S, Behara PK, Madin OC, Hahn DF, Jang H, Gapsys V, Wagner JR, Horton JT, Dotson DL, Thompson MW, Maat J, Gokey T, Wang L-P, Cole DJ, Gilson MK, Chodera JD, Bayly CI, Shirts MR, Mobley DL
Journal of Chemical Theory and Computation 19:3251, 2023 [DOI] [chemRxiv] [GitHub] [examples]
We present a new generation of small molecule force field for molecular design from the Open Force Field Initiative fit to both quantum chemical and experimental liquid mixture data
MEN1 mutations mediate clinical resistance to menin inhibition
/
Perner F, Stein EM, Wenge DV, Singh S, Kim J, Apazidis A, Rahnamoun H, Anand D, Marinaccio C, Hatton C, Wen Y, Stone RM, Schaller D, Mowla S, Xiao W, Gamlen HA, Stonestrom AJ, Persaud S, Ener E, Cutler JA, Doench JG, McGeehan GM, Volkamer A, Chodera JD, Nowak RP, Fischer ES, Levine RL, Armstrong SA, Cai SF
Nature 615:913, 2023 [DOI]
We describe how mutants that confer therapeutic resistance to menin inhibition impact small molecule binding but not interactions with the natural ligand MLL1.
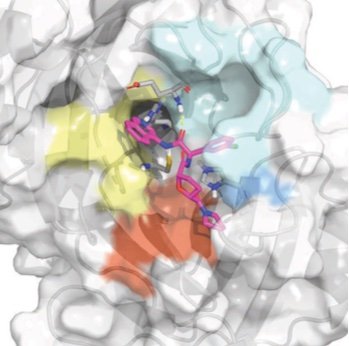
Turning high-throughput structural biology into predictive inhibitor design
/
Saar KL, McCorkindale W, Fearon D, Boby M, Barr H, Ben-Shmuel A, COVID Moonshot Consortium, London N, von Delft F, Chodera JD, Lee AA
PNAS 120:e2214168120, 2023 [DOI]
We demonstrate how potent inhibitors can be predicted from high-throughput structural biology, demonstrating this approach against the SARS-CoV-2 main viral protease (Mpro).
Spatial attention kinetic network with E(n) equivariance
/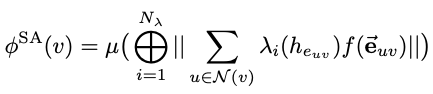
Yuanqing Wang and John D. Chodera
preprint: [arXiv] [code]
This work descibes Spatial Attention Kinetic Networks (SAKE), a new E(n)-equivariant architecture that uses spatial attention, enabling the construction of extremely performant but still accurate machine learning potentials, as well as flows capable of prediction dynamics.
SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials
/
Eastman P, Behara PK, Dotson DL, Galvelis R, Herr JE, Horton JT, Mao Y, Chodera JD, Pritchard BP, Wang Y, De Fabritiis G, and Markland TE
Scientific Data 10:11, 2023 [DOI]
To remedy the lack of large, open quantum chemical datasets for training accurate general machine learning potentials and molecular mechanics force fields for druglike small molecules and biomolecules, we produce the open SPICE dataset, and show how it can be used to build extremely accurate machine learning potentials.
Open Force Field BespokeFit: Automating Bespoke Torsion Parametrization at Scale
/
Horton JT, Boothroyd S, Wagner W, Mitchell JA, Gokey T, Dotson DL, Behara PK, Ramaswamy VK, Mackey M, Chodera JD, Anwar J, Mobley DL, and Cole DJ
Journal of Chemical Informatics and Modeling 62:22, 2022 [DOI]
We describe an automated pipeline for generating tailored force field parameters for small molecules using quantum chemical or quantum machine learning potentials.
End-to-end differentiable molecular mechanics force field construction
/
Yuanqing Wang, Josh Fass, and John D. Chodera
Chemical Science 13:12016, 2022 [DOI] [arXiv] [pytorch code] [JAX code]
Molecular mechanics force fields have been a workhorse for computational chemistry and drug discovery. Here, we propose a new approach to force field parameterization in which graph convolutional networks are used to perceive chemical environments and assign molecular mechanics (MM) force field parameters. The entire process of chemical perception and parameter assignment is differentiable end-to-end with respect to model parameters, allowing new force fields to be easily constructed from MM or QM force fields, extended, and applied to arbitrary biomolecules.
Improving force field accuracy by training against condensed-phase mixture properties
/
Boothroyd S, Madin OC, Mobley DL, Wang L-P, Chodera JD, and Shirts MR
Journal of Chemical Theory and Computation 18:3577, 2022 [DOI] [GitHub]
We use a new automated framework for physical property evaluation and fitting to show how molecular mechanics force fields can be systematically improved by fitting to condensed phase properties.
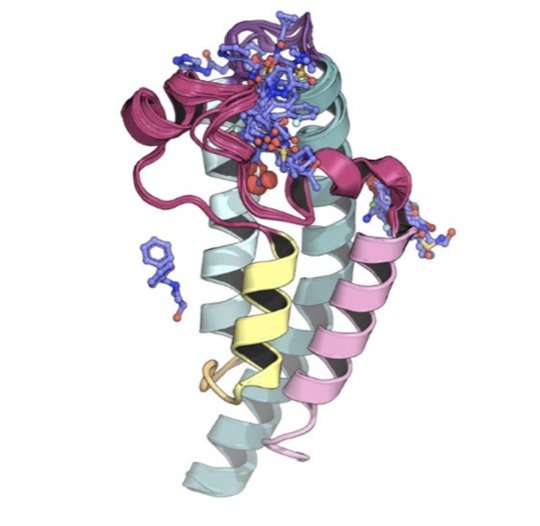
SAMPL7 protein-ligand challenge: A community-wide evaluation of computational methods against fragment screening and pose-prediction
/
Grosjean H, Isik M, Aimon A, Mobley D, Chodera JD, von Delft F, and Biggin PC
Journal of Computer-Aided Molecular Design 36:291, 2022 [DOI]
We field a blind community challenge to assess how well state of the art computational chemistry methods can predict the binding modes of small druglike fragments to a protein target for which no chemical matter is known, PHIP2, using fragment screening at the Diamond Light Source.
CACHE (Critical Assessment of Computational Hit-finding Experiments): A public-private partnership benchmarking initiative to enable the development of computational methods for hit-finding
/
Ackloo S, Al-awar R, Amaro RE, Arrowsmith CH, Azevedo H, Batey RA, Bengio Y, Betz UAK, Bologa CG, Chodera JD, Cornell WD, Dunham I, Ecker GF, Edfeldt K, Edwards AM, Gilsom MK, Gordijo CR, Hessler G, Hillisch A, Hogner A, Irwin JJ, Jansen JM, Kuhn D, Leach AR, Lee AA, Lessel U, Moult J, Muegge I, Oprea TI, Perry BG, Riley, Singh Saikantendu K, Santhakumar V, Schapira M, Scholten C, Todd MH, Vedadi M, Volkamer A, and Wilson TM
Nature Reviews Chemistry 6:287, 2022 [DOI]
We describe CACHE: A new public-private partnership that aims to transform computer-aided drug discovery much the way that CASP transformed protein structure prediction into a reproducible, accurate engineering discipline.
GCN2 kinase activation by ATP-competitive kinase inhibitors
/
Mellinghoff I, Tang CP, Clark O, Ferrarone J, Campos C, Lalani AS, Chodera JD, Intlekofer AM, and Elemento O
Nature Chemical Biology}, 18:207, 2022 [DOI]
We describe paradoxical activation of GCN2 kinase activity by the kinase inhibitor neratinib, and propose a model for how inhibitor-induced dimerization might cause this unusual activity.
Quantum chemistry common driver and databases (QCDB) and quantum chemistry engine (QCEngine): Automation and interoperability among computational chemistry programs
/
Smith DGA, Lolinco AT, Glick ZL, Lee J, Alenaizan A, Barnes TA, Borca CH, Di Remigio R, Dotson DL, Ehlert S, Heide AG, Herbst MF, Hermann J, Hicks CB, Horton JT, Hurtado AG, Kraus P, Kruse P, Lee SJR, Misiewicz JP, Naden LN, Ramezanghorbani F, Scheurer M, Shriber JB, Simmonett AC, Steinmetzer J, Wagner JR, Ward L, Welborn M, Altarawy D, Anwar J, Chodera JD, Dreuw A, Kulik HJ, Liu F, Martinez TJ, Matthews DA, Schaefer III HF, Sponer J, Turney JM, Wang L-P, De Silva N, King RA, Stanton JF, Gordon MS, Windus TL, Sherrill CD, Burns LA
Journal of Chemical Physics} 155:204801, 2021 [DOI]
We describe a new community-wide approach to interoperability for quantum chemistry packages that will enable large-scale applications such as next-generation machine learning for chemistry and automated force field construction for drug discovery.
Teaching free energy calculations to learn from experimental data
/
Marcus Wieder, Josh Fass, and John Chodera
[bioRxiv] [code] [data]
We show, for the first time, how alchemical free energy calculations can be used to not only compute free energy differences between small molecules involving covalent bond rearrangements in systems treated entirely with quantum machine learning potentials, but that these calculations have the capacity to learn to efficiently generalize from conditioning on experimental free energy data.
The Open Force Field Evaluator: An automated, efficient, and scalable framework for the estimation of physical properties from molecular simulation
/
Simon Boothroyd, Lee-Ping Wang, David L. Mobley, John D. Chodera, and Michael R. Shirts
Preprint ahead of submission: [ChemRxiv]
We describe a new software framework for automated evaluation of physical properties for the benchmarking and optimization of small molecule force fields according to best practices.
Mutation in Abl kinase with altered drug binding kinetics indicates a novel mechanism of imatinib resistance
/
Agatha Lyczek, Benedict Tilman Berger, Aziz M Rangwala, YiTing Paung, Jessica Tom, Hannah Philipose, Jiaye Guo, Steven K Albanese, Matthew B Robers, Stefan Knapp, John D Chodera, Markus A Seeliger
Preprint ahead of publication: [bioRxiv]
Here, we characterize the biophysical mechanisms underlying mutants of Abl kinase associated with clinical drug resistance to targeted cancer therapies. We uncover a surprising novel mechanism of mutational resistance to kinase inhibitor therapy in which the off-rate for inhibitor unbinding is increased without affecting inhibitor affinity.
A white-knuckle ride of open COVID drug discovery
/
Frank von Delft, Mark Calmiano, John Chodera, Ed Griffen, Alpha Lee, Nir London, Tatiana Matviuk, Ben Perry, Matt Robinson, and Annette von Delft.
Nature 594:330, 2021.
[DOI] [PDF]
The COVID Moonshot is an open science effort to discover a direct-acting SARS-CoV-2 oral antiviral. Here, we share lessons from this effort, including the missed opportunity to develop a phase 2 ready drug more than a decade ago that could have halted the COVID-19 pandemic in its tracks.