Towards chemical accuracy for alchemical free energy calculations with hybrid physics-based machine learning / molecular mechanics potentials
/
Dominic A. Rufa, Hannah E. Bruce Macdonald, Josh Fass, Marcus Wieder, Patrick B. Grinaway, Adrian E. Roitberg, Olexandr Isayev, and John D. Chodera.
Preprint ahead of submission.
[bioRxiv] [GitHub]
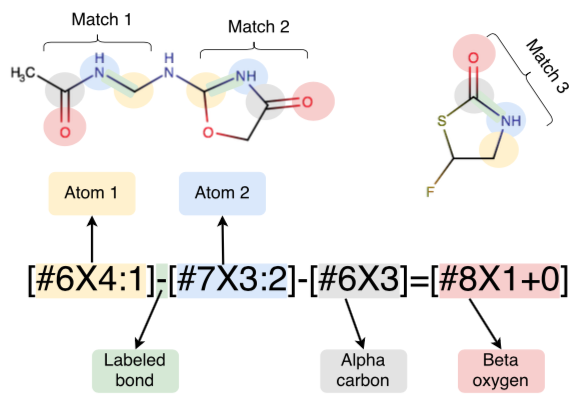
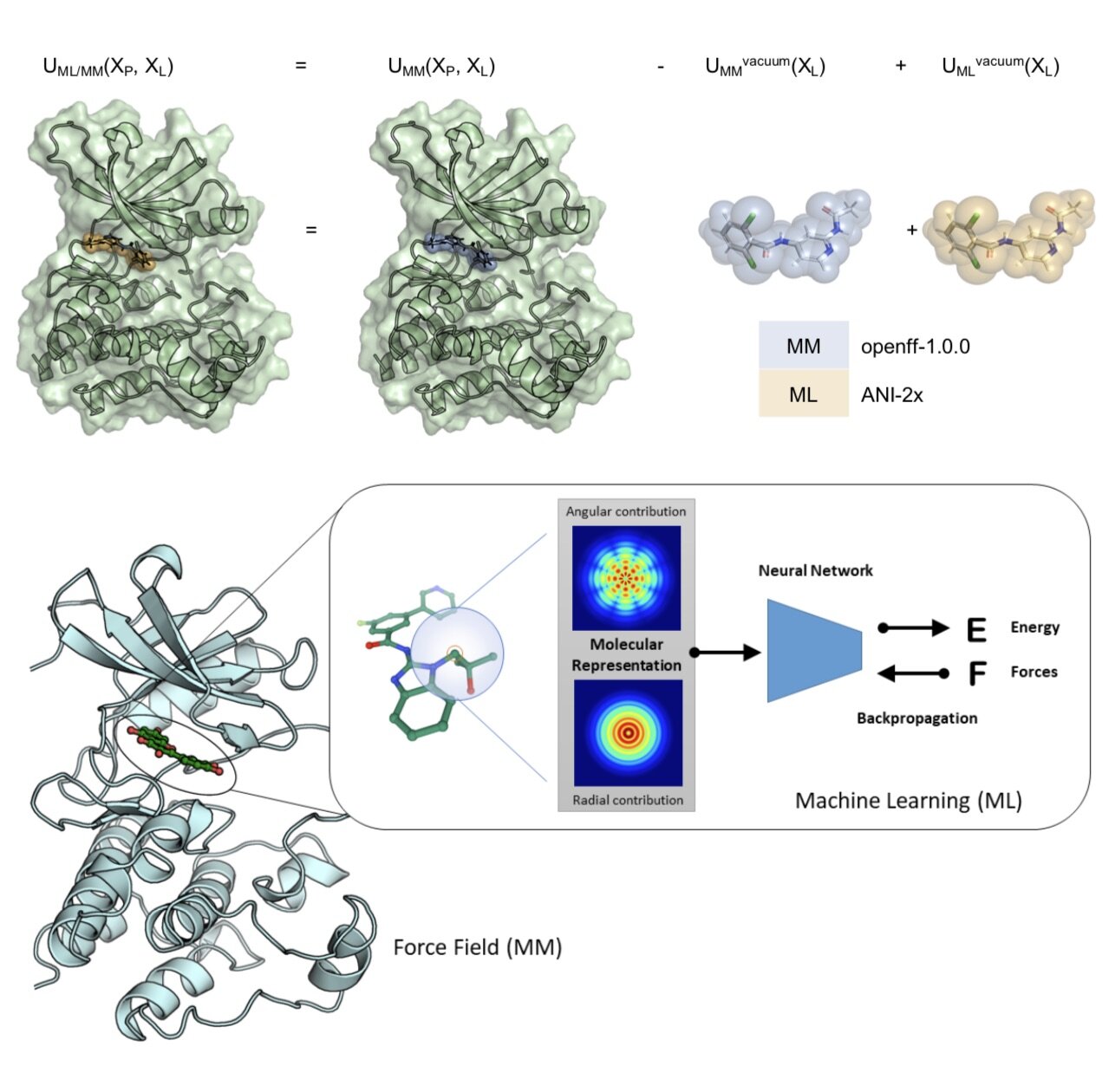
In this first use of hybrid machine learning / molecular mechanics (ML/MM) potentials for alchemical free energy calculations, we demonstrate how the improved modeling of intramolecular ligand energetics offered by the quantum machine learning potential ANI-2x can significantly improve the accuracy in predicting kinase inhibitor binding free energy by reducing the error from 0.97~kcal/mol to 0.47~kcal/mol, which could drastically reduce the number of compounds that must be synthesized in lead optimization campaigns for minimal additional computational cost.